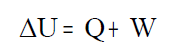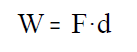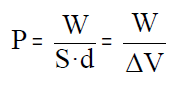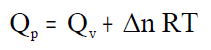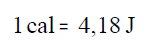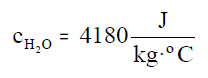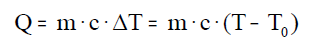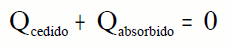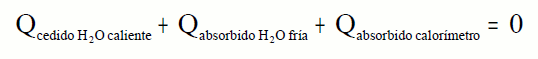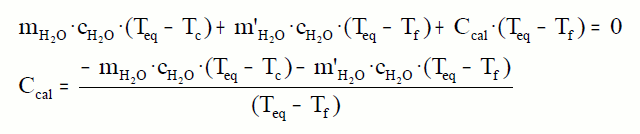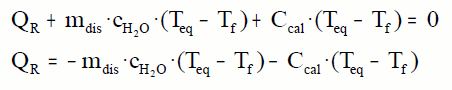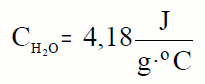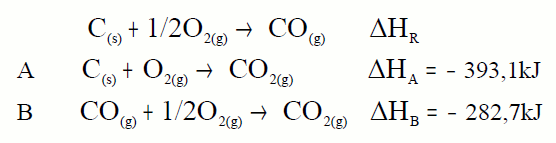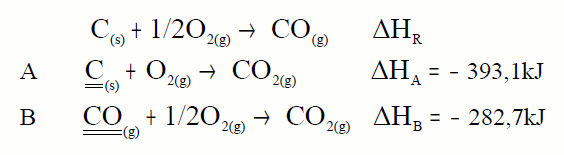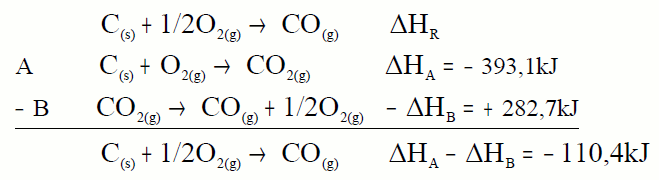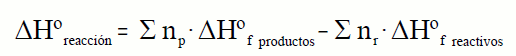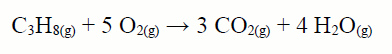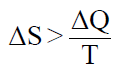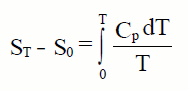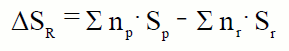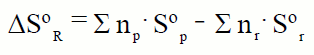|
|
|
|
OBJETIVO DE LA TERMOQUÍMICA
|
| La Termoquímica es la aplicación de la
Termodinámica al estudio de la química. La Termoquímica estudia los intercambios de energía que acompañan a las reacciones químicas. Es un hecho experimental que en toda reacción química hay una variación de energía, manifestada normalmente por la emisión o absorción de calor. El objetivo de la Termoquímica es el estudio de estas variaciones de energía (de su importancia puede darnos idea el hecho de que no sólo hay muchas reacciones, en especial las de combustión, que tienen como único objetivo el aprovechamiento de la energía desprendida) y también el estudio de la espontaneidad de una reacción.
Se puede decir de una manera muy general que la termodinámica es la ciencia de la energía, y se podría añadir que también se ocupa de la entropía.
La termodinámica está basada, esencialmente en solo dos postulados, conocidos como primero y segundo principios de la termodinámica. Estos axiomas no se deducen, en todo caso los podemos considerar generalizaciones de experiencias. De ellos, por razonamientos lógicos, matemáticos, se derivan relaciones entre las magnitudes macroscópicas, es decir, variables que se refieren a un gran conjunto de partículas (volumen, presión, temperatura, calores específicos, entalpía,...). La validez de estas relaciones se puede comprobar experimentalmente, y con eso la de los principios.
Hace falta subrayar que la termodinámica es una teoría macroscópica, no hace falta tener en cuenta para nada la estructura atómica y molecular de la materia. Esta característica le confiere una gran fiabilidad. Puede ser falsa la existencia de los átomos pero la termodinámica no cambiará sus principios, ya que no se deducen teóricamente sino de la experiencia.
La termodinámica no describe el proceso de cambio de los sistemas ni indica el tiempo necesario para lo mismo, informando solo acerca de la posibilidad de que exista tal cambio. A pesar de todo, saber sí una reacción es posible o no puede ahorrar muchos esfuerzos inútiles, así, si la termodinámica hubiera dicho que era imposible obtener diamantes a partir de grafito, a nadie se le
ocurriría esforzarse intentándolo.
Los objetivos de la termoquímica los podemos resumir así:
- Estudio de las variaciones de energía, como entalpías de reacción, energías libres...
- Saber, fijadas ciertas condiciones (como por ejemplo la temperatura y las concentraciones), si es factible una reacción o no.
- Obtención, teórica, de las constantes de equilibrio y métodos para mejorar los rendimientos de las reacciones.
Con respecto a un proceso en concreto la termoquímica no nos dice nada de su velocidad ni de su
mecanismo, esto será objeto de la cinética química.
 |
|
DEFINICIONES BÁSICAS
|
Sistema es una cantidad de materia que nosotros aislamos, bien sea en la realidad o imaginativamente, para estudiarla. Los sistemas termodinámicos pueden ser:
- Abiertos, en este caso intercambian con los alrededores materia y además energía.
- Cerrados, cuando sólo intercambian energía. Su masa permanece constante, no entra ni sale materia.
- Aislados, cuando no intercambian con los alrededores (el medio ambiente) ni materia ni energía.
Todo sistema se caracteriza macroscópicamente por unos valores concretos de sus propiedades observables experimentalmente, llamadas
variables de estado, como por ejemplo: la presión (P), el volumen (V), la temperatura (T) y la composición (χi). Para una sustancia pura la composición, al ser siempre la misma, no hace falta especificarla. Es suficiente, en este caso, para conocer el estado del sistema con tres variables, P, V, T.
Hay variables que no cambian su valor aunque dividamos el sistema, no dependen de la cantidad de masa, se dice que son
intensivas y otras que sí dependen de la cantidad de masa, les llamamos
extensivas. Son intensivas: presión, temperatura, densidad, composición... y son extensivas: volumen, masa, y otras como energía interna, entalpía, energía libre, entropía...
Cuando se modifica una, o más, variables de estado decimos que el sistema sufrió un proceso de cambio o transformación.
En este curso estudiaremos únicamente sistemas que se encuentran en estado de equilibrio termodinámico. Por suerte, para describir dichos sistemas sólo hace falta conocer un reducido número de variables termodinámicas. Como por ejemplo, si el sistema es un gas llega con conocer la presión, el volumen y la temperatura para describir perfectamente su estado. En general, las variables de estado se encuentran relacionadas entre sí mediante ecuaciones matemáticas; como por ejemplo: P V = n R T. (Ecuación de estado)
Existen algunas variables termodinámicas que tienen la importantísima cualidad de que su valor depende exclusivamente del estado del sistema, y no de las formas intermedias por las que este evoluciona. A estas variables se les denomina
funciones de estado. Son funciones de estado: el volumen, la energía interna, la entropía, la entalpía, la presión, la temperatura, porque su valor, dentro de un instante dado, es independiente del mecanismo que siga el proceso cuando se pasa de un estado inicial a otro final. Las variaciones que experimentan estas funciones sólo dependen del estado inicial y final del sistema, sea cuál sea el camino de transformación.
|
|
PRIMER PRINCIPIO DE LA
TERMODINÁMICA
|
| Este principio refleja la ley de conservación de la energía para un sistema termodinámico, y su objetivo es controlar los intercambios energéticos que tienen lugar entre él y su entorno. |
|
El Primer principio de la
Termodinámica establece que la energía de un sistema se conserva siempre. Si al experimentar un proceso disminuye la energía del sistema debe aparecer una cantidad equivalente de energía en su entorno. |
|
Cuando en un sistema cerrado se produce un aumento o merma de su energía total, implicará necesariamente una merma o aumento, respectivamente, de la misma cantidad en su entorno. De todas las posibles formas de energía que producen o consumen las reacciones químicas (calor, trabajo, energía eléctrica, energía luminosa, etc.) en este tema únicamente se van a considerar el
calor y el trabajo. De hecho, el primer principio de la termodinámica explica como afectan los intercambios de calor y trabajo a la energía de un sistema; para ello se emplea una magnitud denominada
energía interna que se representa por U. Con estos tres conceptos: energía interna (U), trabajo (W) y calor (Q) puede enunciarse el primero principio de la termodinámica cómo:
|
|
"La variación de la energía interna de un sistema es igual al calor desprendido o absorbido por el sistema
más el trabajo realizado por o sobre el sistema" |
|
Criterio de signos.
Para que el enunciado del primer principio sea coherente hace falta especificar la dirección en la que fluye la energía, ya sea en forma de calor o de trabajo. Para ello se adopta un criterio de signos que debe mantenerse siempre.
El criterio propuesto se fundamenta en las siguientes apreciaciones:
Cuando el sistema cede calor al exterior su energía total disminuye, la variación de energía interna debe ser negativa (ΔU<0) y por lo tanto el calor debe ser negativo. Pero si el sistema absorbe calor del entorno su energía total aumenta
(ΔU>0), por consiguiente, este calor debe ser positivo.
Cuando el sistema se expande realiza trabajo sobre su entorno a costa de su energía total, que debe disminuir
(ΔU<0). El trabajo de expansión debe ser negativo. En una compresión, la energía total aumenta
(ΔU>0) y el trabajo de compresión debe ser positivo.
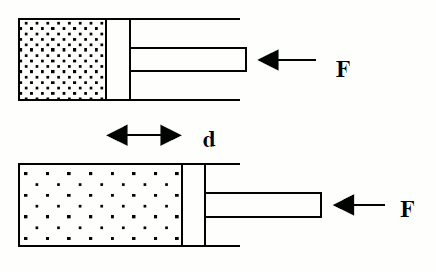
Trabajo de expansión de un gas:
Para mantener un gas a presión en un cilindro hay que realizar una fuerza F. Si la presión interna del gas consigue vencer la fuerza, el gas se expande y realiza un trabajo que vale
donde, F, es la fuerza que se realiza desde el exterior sobre el émbolo para mantener el gas a presión y,
d, es el desplazamiento del émbolo.
La presión del gas en el interior, P, es igual al cociente entre la fuerza,
F, y la superficie, S, del émbolo:
si sustituimos la fuerza de la expresión del trabajo se obtiene:
El producto de la superficie del émbolo por su desplazamiento (S·d) es el cambio de volumen que experimenta el sistema con lo que finalmente podemos escribir:
que es la expresión del trabajo de expansión de un gas. Recuerda que
el trabajo de expansión según nuestro criterio de signos es negativo
pues se realiza a costa de la energía interna del sistema. En esta
expresión V es el volumen final y V0 el volumen inicial.
Expansión ⇒ V>V0 ⇒ − P·(V−V0)<0 ⇒
ΔU<0
Compresión ⇒ V<V0 ⇒ − P·(V −V0)>0 ⇒
ΔU>0
Comentarios sobre la energía interna:
La energía interna de un sistema, U, es la suma de todas las energías contenidas en dicho sistema. Se considera que la energía total de un sistema gaseoso es la suma de las energías cinéticas de sus moléculas:
U = Et + Er + Ev + Ee +
En
Se puede apreciar que hay términos debidos a la energía cinética de las moléculas, energías de traslación, rotación y vibración, y términos debidos a la energía potencial, como la atracción electrostática entre las moléculas, y hasta términos debidos a las fuerzas nucleares.
La energía interna de un sistema (U) es una función de estado, y su valor absoluto es desconocido; el único que se puede hacer es medir su variación cuando en el sistema termodinámico se produzca una transformación. A esta variación se le llamará variación de energía interna
(ΔU).
EJERCICIOS
PARA PRACTICAR
|
|
CALORES DE REACCIÓN.
ENTALPÍA
|
| En una reacción química ΔU representa la diferencia de energía interna entre su estado final (productos) y su estado inicial (reactivos), con lo que el primer principio se puede expresar:
Los valores de Q y W se refieren a los efectos de calor y trabajo que acompañan a una reacción química.
Aunque el primer principio puede aplicarse a una reacción en cualquiera estado, sin limitación alguna, en este caso nos referiremos a dos casos especialmente sencillos: al cálculo de
ΔU cuando el volumen permanece constante (ΔUv) y al cálculo de
ΔU cuando la presión permanece constante (ΔUp), siendo este segundo caso lo que más nos interese.
Transformaciones a volumen constante:
En este caso, como no hay variación de volumen, el trabajo es nulo, con el que la expresión del primer principio queda:
donde Qv es el intercambio de calor a volumen constante. Esta condición se da cuando llevamos a cabo a reacción en una bomba calorimétrica en la que no puede haber contracción o dilatación del sistema. En los procesos a volumen constante, el valor del calor medido en la bomba calorimétrica da directamente la variación de la energía interna, U, para la reacción que se está estudiando.
El calor absorbido o desprendido en una reacción química a volumen constante
(Qv) es igual a la variación de energía interna del sistema.
Si Q>0 ⇒ ΔU>0 ⇒ U aumenta
Si Q<0 ⇒ ΔU<0 ⇒ U disminuye
Transformaciones a presión constante, concepto de entalpía:
La mayoría de las reacciones se realizan a presión constante, ya que en los laboratorios se trabaja a menudo en recipientes abiertos, o lo que es lo mismo, a presión atmosférica. Casi todos los procesos que vamos estudiar son de este tipo.
Para calcular el intercambio de calor asociado a una reacción a presión constante, se emplea también el primer principio. En los procesos a presión constante es frecuente que a medida que transcurre la reacción exista un pequeño cambio de volumen, que producirá un trabajo de expansión:
siendo ΔV = V productos – V reactivos, con lo que el primer principio toma la forma:
donde Qp = variación de calor a presión constante.
Despejando el calor Qp:
Desarrollando esta expresión:
y si se agrupan los términos correspondientes a los estados inicial y final se obtiene:
P·V tiene dimensiones de energía y sumada a la energía interna U nos dará una nueva energía que llamaremos
entalpía y que representaremos por H:
que sustituida en la ecuación anterior nos da el calor intercambiado en una reacción a presión constante:
La entalpía, H, es otra forma de medir la energía de un sistema; H y
H0 son los valores de la energía calorífica del sistema en los estados final e inicial, y su diferencia,
ΔH, representa el calor transferido por el sistema en cualquiera proceso que tenga lugar a presión
constante.
La entalpía es una función de estado, e igual que le ocurría a la energía interna, no se pode medir su valor absoluto, sólo se
puede medir su variación (en procesos a presión constante).
Si H>H0 ⇒ ΔH>0 ⇒ Qp>0
el sistema absorbe calor, luego el proceso es endotérmico.
Si H<H0 ⇒ ΔH<0 ⇒ Qp<0
el sistema desprende calor, luego el proceso es exotérmico.
|
|
ENTALPÍA DE REACCIÓN.
DIAGRAMAS ENTÁLPICOS
|
| Como la entalpía es una función de estado, cuando un sistema evoluciona desde un estado inicial a otro final, su variación de entalpía se expresa cómo:
En el caso de una reacción química el estado inicial son los reactivos y el estado final son los productos, por lo tanto la variación de entalpía de una reacción será:
Si Hp>Hr ⇒ ΔH>0
⇒ Qp>0, (el sistema absorbe calor) la reacción es endotérmica.
Si Hp<Hr ⇒ ΔH<0 ⇒
Qp<0, (el sistema desprende calor) la reacción es exotérmica.
La variación de entalpía de una reacción se puede expresar
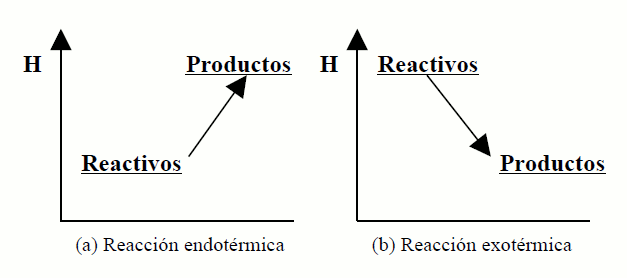
gráficamente con ayuda de los diagramas entálpicos, en los que se representa sobre una escala de energías la entalpía de reactivos y productos. Al no poderse conocer el valor absoluto, la escala de entalpías (en ordenadas) tiene un origen arbitrario. La figura (a) representa el diagrama entálpico para una reacción endotérmica, y la (b) para una reacción exotérmica.
Ecuaciones termoquímicas:
En termoquímica, para representar una reacción, además de indicar los coeficientes estequiométricos se indica el estado (sólido, líquido o gas) de cada una de las substancias. También debe incluirse el intercambio de calor (ganancia o pérdida) que tiene lugar en el proceso. Esta forma de representar la ecuación recibe el nombre de
ecuación termoquímica. La inclusión del calor intercambiado en el proceso se pode hacer de dos maneras: poniendo explícitamente el calor, o poniendo el valor que para ese proceso tiene ΔH o ΔU. En este último caso podemos saber, además, si el proceso ocurre a presión o a volumen constante. Ejemplo:
|
|
MEDIDAS CALORIMÉTRICAS
|
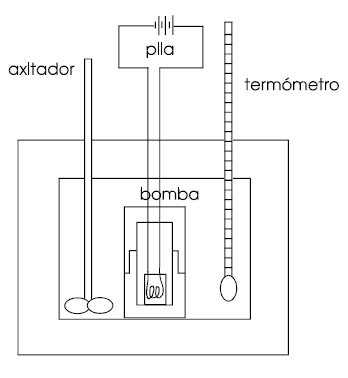
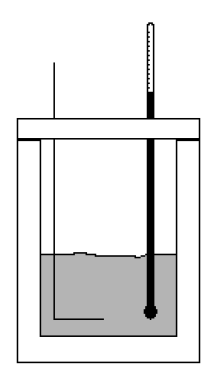
| Las reacciones químicas pueden realizarse dentro de un recipiente herméticamente cerrado, indeformable mecánicamente, de paredes rígidas que impidan cualquier variación de volumen del sistema en el transcurso de las mismas. Si las paredes del recipiente permiten el paso libre de calor a través del mismo, la cantidad de calor intercambiado cuando tiene lugar la reacción química se llama calor de reacción a volumen constante.
Estos calores de reacción a volumen constante se miden mediante dispositivos como el de la figura llamados bomba calorimétrica. Para medir un calor de combustión la muestra pesada se introduce en un crisol (que se introduce a su vez en la bomba) y se quema completamente en oxígeno a presión. La muestra se calienta mediante un filamento de ignición de hierro, que se pone incandescente cuando se pasa una corriente eléctrica procedente de una batería. El calorímetro está aislado mediante un manto aislante térmico. La temperatura del fluído calorimétrico se mide con el termómetro. A partir del cambio de temperatura y habida cuenta la cantidad de calor añadido a través del filamento, se puede calcular el calor de reacción.
Este mismo proceso se puede realizar en un recipiente dotado de una pared móvil, sobre la que actúa una presión constante (en general la atmosférica). Después de restablecida la temperatura inicial, terminada la reacción, a veces se observa que el volumen inicial no es igual al volumen final, pues el número de moles de gas de los reactivos no tiene que coincidir con el número de moles de gas de los productos. Suponiendo comportamiento ideal de los gases que participan en la reacción podemos establecer que:
¿Cómo podemos hacer medidas calorimétricas en un calorímetro?
Primero recordaremos como medir el calor:
Hagamos un experimento: vamos a aportar calor a una cierta cantidad de agua.
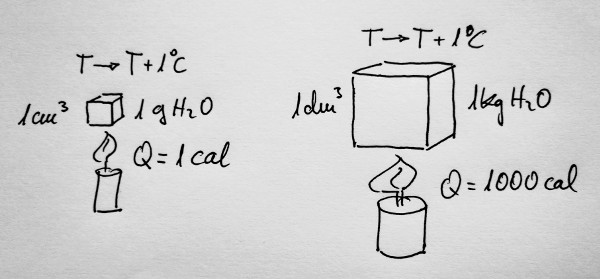
Si aportamos calor a un gramo de agua para que su
temperatura aumente un grado centígrado, o un grado Kelvin, a esa
cantidad de calor se le llamó caloría. Y se representa por cal.
De la misma forma, si aportamos calor a un kilogramo de
agua para que su temperatura aumente un grado centígrado, o un grado
Kelvin, esa cantidad de calor serán 1000 calorías.
Pero la caloría no es la unidad de energía en el Sistema
Internacional de unidades, esa unidad es el Julio. La equivalencia
entre julios y calorías es:
Por tanto, el calor que tenemos que aportar a 1kg de agua
para que su temperatura aumente 1ºC, o 1K, es 4180J. Esta cantidad se
conoce como el calor específico del agua.
Es un valor que podemos determinar para cada sustancia
pura y es de gran utilidad para medir calores.
Calor específico de una sustancia, c, es el
calor que hay que proporcionar a una unidad de masa, 1kg, para que su temperatura aumente
1K.
Su unidad es J/kg K. Por ejemplo, el calor específico del agua es
4180J/kg K, ya que hay que aportar 4180J a un kilogramo de agua para que su temperatura
aumente un Kelvin, o un grado centígrado.
|
Sustancia |
Calor
específico, c (J/kg K) |
Sustancia |
Calor
específico, c (J/kg K) |
| Agua (liq.) |
4180 |
Oro |
129 |
| Hielo |
2114 |
Plata |
237 |
| Vapor de agua |
2080 |
Mercurio |
139 |
| Etanol |
2440 |
Cobre |
385 |
| Amoníaco (liq.) |
4700 |
Hidrógeno |
14300 |
| Aluminio |
897 |
Nitrógeno |
1040 |
| C (grafito) |
710 |
Oxígeno |
918 |
| Hierro |
450 |
Arena |
835 |
| Plomo |
129 |
Granito |
790 |
El calor absorbido o cedido por un cuerpo se calcula con
la siguiente ecuación:
Donde (Q) es el calor cedido o absorbido, (m) la masa, (c)
el calor específico , (T) la temperatura final, y (To) la temperatura
inicial
Un calorímetro es un aparato que nos va a permitir calcular calores de reacción, que por ser a presión constante serán entalpías. Su construcción debe garantizar que el calor no salga del mismo, aunque el mismo pueda absorber cierta cantidad de calor.
En un calorímetro:
Pues se conserva la energía.
El calor cedido o absorbido por una sustancia lo podemos calcular a partir de
donde m es la masa, c el calor específico y ΔT la variación de la temperatura.
El calorímetro también absorberá una cierta cantidad de calor que debemos calcular para cada calorímetro.
donde Ccal es la capacidad calorífica del calorímetro.
¿Cómo determinar la capacidad calorífica del calorímetro?
Introducimos una cierta cantidad de agua en el calorímetro a temperatura ambiente, el calorímetro
también estará a esta temperatura, que llamaremos Tf. Calentamos otra cantidad de agua y medimos su temperatura, que llamaremos
Tc. Introducimos rápidamente esta cantidad de agua en el calorímetro.
Medimos la temperatura que llamaremos Teq. El calor cedido por el agua caliente debe coincidir con el calor absorbido por el agua fría y el calorímetro.
Este valor lo conservaremos para todas las experiencias con ese calorímetro.
¿Cómo determinar un calor de disolución?
Añadimos una cantidad de agua al calorímetro y medimos la temperatura. Luego medimos una masa de sal y la añadimos al calorímetro. Agitamos hasta la disolución completa. Medimos la temperatura de equilibrio y realizamos los cálculos:
El calor específico de la disolución podemos asimilarlo al calor específico de agua sin cometer mucho error para disoluciones diluidas.
Práctica de calorimetría: Medida de calores de reacción, y
comprobación de la ley de Hess:
EJERCICIOS
PARA PRACTICAR
|
|
ENTALPÍA DE REACCIÓN
|
| La variación de entalpía que se produce durante una reacción a presión constante se puede expresar como la diferencia entre las entalpías de los productos y las entalpías de los reactivos.
Pero no podemos conocer las energías absolutas asociadas a cada sustancia. Ya lo vimos también para el caso de la energía interna. ¿Cómo calcular entonces las entalpías de las reacciones? Tenemos cuatro formas de hacerlo:
1. Mediante medidas calorimétricas.
2. Mediante la ley de Hess.
3. Mediante entalpías de formación.
4. Mediante entalpías de enlace.
|
|
LEY DE HESS
|
| La entalpía es una función de estado y su variación ΔH a presión constante solo depende de los estados inicial y final, pero no de los intermedios por los que transcurre la reacción.
Basándose en esta propiedad, el suizo Germain Henri Hess formuló en 1840 el principio que lleva su nombre:
|
|
Ley de Hess: Cuando una reacción química puede expresarse como suma algebraica de otras, su calor de reacción es igual a la suma algebraica de los calores de las reacciones parciales. |
|
En muchos casos no es posible determinar experimentalmente la entalpía de reacción de un proceso; como por ejemplo la entalpía de formación del CO, no es medible en un calorímetro. En estos casos la ley de Hess es especialmente útil:
A partir de estas dos reacciones podemos calcular el calor de la primera. Tendremos que colocarlas de forma que sumadas nos den la que buscamos, lo mismo que hacemos con las ecuaciones lo hacemos con los calores. ¿Cómo colocarlas? Buscamos en
cada ecuación la sustancia clave que es la sustancia de la
ecuación problema que aparece en exclusiva en cada ecuación, esa sustancia la colocamos en la misma posición y con el mismo
coeficiente que en la ecuación problema:
La ecuación A la dejamos tal cual, pues el carbono aparece como
reactivo y con coeficiente 1. Pero la B la invertimos pues el CO aparece como reactivo y en la ecuación problema como producto aunque con el mismo coeficiente. Lo mismo que hacemos con las ecuaciones lo hacemos con los calores.
Sumamos y comprobamos que nos da la ecuación problema, lo mismo hacemos con los calores.
EJERCICIOS
PARA PRACTICAR
|
|
ENTALPÍA NORMAL DE
FORMACIÓN
|
| Otra forma de poder calcular la entalpía de una reacción podría ser tabulando los valores de la entalpía de cualquier reacción. Pero como te puedes imaginar esto es poco menos que misión imposible. Si se tuviera que tabular la ΔH para todas y cada una de las miles de reacciones que se pueden realizar a presión constante, seguramente no encontraríamos sitio para almacenar tanta información.
Un compuesto determinado puede intervenir en miles de reacciones químicas pero sólo se tabula una de ellas; aquella en la que se forma este compuesto a partir de sus elementos en el estado de agregación (sólido, líquido o gas) en el que se encuentran en las condiciones habituales de laboratorio (condiciones estándar): presión de 1atm y temperatura de 298K (25ºC).
A las entalpías de los elementos, en el estado de agregación más estable en las condiciones anteriores, se les asigna el valor cero por la propia definición de entalpía de formación.
La entalpía normal de formación de un compuesto en condiciones estándar, también denominada calor de formación, se representará por ΔHºf, y se define como: el cambio de entalpía, ΔH, que tiene lugar cuando se forma un mol de compuesto a partir de los elementos que lo constituyen en sus estados de agregación más estables en condiciones estándar.
La entalpía es una forma de energía, por lo tanto es una magnitud extensiva que depende de la masa del sistema; por esta razón, al definir la entalpía de formación se especifica que se refiere a la formación de un mol de compuesto. Ejemplos:
Ag(s) + 1/2 Cl2(g) → AgCl(s) ΔH = −
127 kJ/mol
1/2N2(g) + 1/2 O2(g) → NO(g) ΔH = 90,4 kJ/mol
1/2 N2(g) + 3/2 H2(g) → NH3(g) ΔH = −
46,0 kJ/mol
Si observas la mayoría de los calores de formación en condiciones estándar en una tabla comprobarás que son negativos, por lo que dichos procesos serán exotérmicos, y las descomposiciones de los compuestos serán, por consiguiente,
endotérmicas.
TABLA
DE DATOS QUÍMICOS. En esta tabla puedes encontrar
tabuladas las entalpías de formación de muchas sustancias.
|
|
ENTALPÍA DE REACCIÓN A
PARTIR DE ENTALPÍAS DE
FORMACIÓN
|
| Como se indicó anteriormente de forma genérica la variación de la entalpía de una reacción es:
Ahora estamos capacitados para realizar el cálculo de la entalpía de una reacción en condiciones estándar de forma exacta, ya que aunque no conocemos las entalpías absolutas si podemos asociar a cada sustancia su entalpía de formación en las condiciones estándar.
Las entalpías de formación en dichas condiciones corresponden a la formación de un mol de sustancia; por eso, al ser la entalpía una magnitud extensiva, cuando en la ecuación ajustada los coeficientes de las sustancias que intervienen en la reacción sean diferentes de 1, será preciso multiplicar las entalpías de formación por los coeficientes estequiométricos para calcular la entalpía de la reacción. La entalpía de reacción en condiciones estándar se puede escribir como:
donde: np y nr son los coeficientes estequiométricos de los productos y de los reactivos.
Esta ecuación es una aplicación de que la entalpía es una función de estado, y por lo tanto de la ley de Hess.
EJEMPLO 1: Calcula la entalpía normal para la reacción de combustión del etanol
C2H5OH(l)
Ajustamos la reacción: C2H5OH(l) + 3
O2(g) → 2 CO2(g) + 3 H2O(l)
Dado que: ΔHºR = Σ np·ΔHºfp − Σ
nr·ΔHºfr
y que ΔHºf [O2(g)] = 0
utilizando la
tabla
de datos químicos.
ΔHºR = 2mol·ΔHºf [CO2(g)] + 3mol·ΔHºf
[H2O(l)] − 1mol·ΔHºf [C2H5OH(l)] =
= 2mol (− 393,7kJ/mol) + 3mol (− 285kJ/mol) − 1mol (− 277kJ/mol) = − 1365,4 kJ
EJERCICIOS
PARA PRACTICAR
|
|
LAS ENERGÍAS DE LOS ENLACES Y SU RELACIÓN CON EL CALOR DE REACCIÓN
|
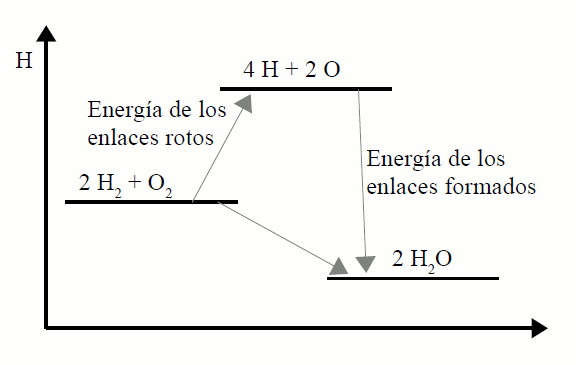
| A veces no se conocen las entalpías de formación de determinados compuestos. Un método aproximado para calcular calores de reacción, sabiendo que en una reacción química se rompen unos enlaces y se forman otros nuevos, es asignar a cada enlace la energía que necesitamos para romperlos.
La energía de enlace o entalpía de enlace se define como: el flujo de energía cuando se rompe un mol de enlaces en estado gaseoso a presión constante.
La entalpía de enlace siempre es positiva, ya que es la energía que absorbe la molécula cuando rompe uno de sus enlaces químicos.
El concepto de energía de enlace nos ayuda a entender por que algunas reacciones son exotérmicas y otras endotérmicas, pues si los enlaces de las moléculas de los productos son más fuertes que los enlaces de las moléculas reaccionantes, la reacción será exotérmica, ya que:
En el caso de que se pase de enlaces débiles en los reactivos a enlaces más fuertes en los productos, se producirá un desprendimiento de energía y ΔH será negativa, por lo que la reacción será exotérmica.
Los valores de las entalpías de enlace son valores medios calculados a partir de diferentes compuestos. Por lo tanto serán valores aproximados, aunque siempre será mejor tener un valor aproximado de una magnitud que no tener ninguno.
El cálculo de las entalpías de reacción a partir de las entalpías de enlace es especialmente interesante en los compuestos orgánicos, debido al inmenso número de estos que hace que no se disponga de las entalpías de formación de muchos compuestos, además de
poseer muy pocos tipos de enlace, así que con una tabla pequeña de entalpías de enlace tabuladas podemos calcular muchísimas entalpías de reacción. Para el cálculo será fundamental conocer los enlaces que se dan en los compuestos, para lo cual ayuda conocer las formulas desarrolladas de los compuestos que forman la reacción.
EJEMPLO 2: Calcula la ΔHcombustión a 298K del propano CH3‒CH2‒CH3 a partir de los siguientes datos: ΔHº(C‒C)=348, ΔHº(C‒H)=413, ΔHº(O‒H)=463, ΔHº(O=O)=495, ΔHº(C=O)=802 todos los valores en kJ/mol
ΔHºR = 8mol · ΔHº(C‒H) + 2mol · ΔHº(C‒C) + 5mol
· ΔHº(O=O) ‒ 6mol · ΔHº(C=O) ‒ 8mol · ΔHº(O‒H) =
= 8mol · 413kJ/mol + 2mol · 348kJ/mol + 5mol · 495kJ/mol ‒ 6mol ·
802kJ/mol ‒ 8mol · 463kJ/mol = ‒2041kJ
Cuando utilizamos entalpías de formación el resultado es de ‒2044,5 kJ, es un resultado aproximado,
pero será de gran utilidad cuando no tengamos datos de entalpías de formación, o no podamos calcular experimentalmente dicha entalpía.
EJERCICIOS
PARA PRACTICAR
|
|
ESPONTANEIDAD.
CONCEPTO DE ENTROPÍA.
|
| Teniendo en cuenta lo estudiado anteriormente podemos llegar a la siguiente conclusión: desde el punto de vista energético, existen dos tipos de reacciones principales:
a) Las que desprenden calor al formarse los productos a partir de los reactivos mediante una transformación química, es decir, son
exotérmicas y, por lo tanto,
energéticamente favorables.
b) Aquellas en las que necesitamos un aporte continuo del exterior para que se puedan realizar, pues nunca se alcanza un mínimo energético, y en principio no serán favorables desde este punto de vista. Son
endotérmicas.
Ahora bien, parece que el factor energético no es el único que interviene en la espontaneidad de una reacción, pues existen en la naturaleza
procesos
energéticamente desfavorables (endotérmicos), que tienen lugar de una manera espontánea, como por ejemplo la fusión del hielo y la evaporación del agua. De igual manera, la disolución de la sal común en agua es un proceso endotérmico que transcurre espontáneamente:
NaCl(s) + H2O(l) → Cl–(aq) +
Na+(aq) ; ΔH = 3,9 kJ
También se descompone espontáneamente el carbonato de amonio en
hidrogenocarbonato de amonio y amoníaco:
(NH4)2CO3 → (NH4)HCO3 +
NH3 ; ΔH = 40 kJ
Por lo tanto, el factor energético no será el único que deba tenerse en cuenta cuando se trate de predecir si una reacción química va a tener lugar de manera espontánea o no.
Para comprender algunas de las condiciones que determinan si un proceso puede o no realizarse, es útil observar algunas transformaciones que se realizan espontáneamente sin aporte energético exterior. Ejemplos de estas transformaciones son:
-
la expansión de un gas en un espacio vacío
-
la difusión de dos gases dentro de un recipiente hasta que la mezcla sea homogénea
-
la disolución de un sólido en agua
-
la conductividad de calor a lo largo de una barra de metal que tiene un extremo caliente y el otro frío
Una característica fundamental de estos procesos es que no se invierten por sí solos. Para que se inviertan se requiere la acción de un agente externo, es decir, los procesos espontáneos no son
termodinámicamente reversibles (son procesos irreversibles); el conocimiento de este hecho experimental fue la base del
segundo principio de la Termodinámica.
La historia de la segunda ley de la Termodinámica empieza
con el francés Sadi Carnot. Carnot estaba interesado en la eficiencia
de los motores térmicos. Su motor extraía calor de un foco caliente y cedía
calor a un foco frío, produciendo una cierta cantidad de trabajo. La
eficiencia del motor ideal de Carnot no podía ser igualada o superada por
ningún motor real.
Estas ideas de Carnot fueron la base para que el inglés
William Thomson y el alemán Rudolf Clausius formularan el Segundo
principio de la Termodinámica.
La primera formulación del segundo principio se la debemos a
Kelvin.
|
|
No puede haber ningún motor cíclico
cuyo único efecto sea extraer energía de un reservorio de calor y
convertirla completamente en trabajo. |
|
Por tanto establece que, aunque el calor es una forma de
energía, es imposible convertir completamente calor (o energía térmica) en
trabajo. Aunque lo contrario sí es posible, se puede convertir del todo
trabajo en calor.
La formulación de Clausius es una evidencia que tenemos
todos,
|
|
El calor siempre fluye de un cuerpo
más caliente (a más temperatura) a un cuerpo más frío (a menos
temperatura). |
|
Nunca se observa el proceso inverso de forma espontánea. De
la misma forma que el calor siempre fluye en un sentido ya vimos antes que
otros procesos irreversibles también tenían lugar siempre en un determinado
sentido. ¿Pero por qué? Es Clausius quien propone que hay una magnitud que
rige estos procesos y que llamó entropía, S. La entropía, es función de estado
y extensiva. ¿Pero qué mide realmente la entropía?
Procesos irreversibles: En la naturaleza hay procesos que partiendo de una situación inicial transcurren en un sentido, sin que se pueda recuperar la situación inicial, a no ser que intervenga un agente externo. Si se interconectan dos recipientes que contienen gases diferentes, al transcurrir el tiempo y sin necesidad de ninguna acción exterior, los gases se combinan hasta convertirse en una mezcla homogénea. Por el contrario, dos gases que formen una mezcla homogénea nunca se van a separar, por mucho tiempo que se deje evolucionar el sistema.
Procesos reversibles: Otros procesos transcurren a través de una sucesión de estados de equilibrio, de suerte que en cualquier instante se pueda invertir el proceso y hacerlo evolucionar en sentido contrario hasta el estado inicial.
Según Clausius el segundo principio de la Termodinámica lo
podemos formular de la siguiente manera:
|
|
El Segundo principio de la Termodinámica:
"En un proceso reversible la entropía es constante pero, si es irreversible, la entropía aumenta" |
|
En física estábamos acostumbrados a que en los procesos de
cambio regían leyes de conservación, conservación de la masa, de la
energía, del momento, de la carga, pero una magnitud que aumenta siempre
no es fácil de entender.
Universo Mecánico 47: Entropía.
Para entender de forma cualitativa el concepto de entropía
se acude a veces al concepto de orden y desorden. Esta no es la mejor
forma de acercarnos al concepto de entropía pero como primera
aproximación puede ser válida.
¿Qué entendemos por orden y desorden? Lo que puede ser orden
para una persona puede no serlo para otra. Se dice a veces que los cuerpos muy desordenados tienen una elevada entropía; por el contrario, los cuerpos o sistemas muy ordenados poseen bajo contenido entrópico. Esto hace que, cuando un sólido funde, se evapora o se disuelve, sus moléculas aumentan la capacidad de moverse (aumentan los grados de libertad), diremos que están más desordenadas, o lo que es equivalente, su entropía aumenta.
Intentaremos aclarar más adelante estos conceptos.
En un sistema aislado, que no puede intercambiar con el entorno ni materia ni energía, es posible un cambio si ello supone un aumento de entropía; sin embargo, la evolución del sistema se detiene cuando la entropía es máxima.
Un sistema cerrado, que no intercambia materia con el entorno, pero sí energía, puede evolucionar de dos maneras:
a) Mediante un proceso reversible en el cual la entropía ganada o perdida por el sistema debe coincidir con la perdida o ganada por el entorno:
ΔSsistema = – ΔSentorno
ΔStotal = 0 (Proceso isoentrópico)
b) Mediante un proceso irreversible en el cual se incluyen la mayoría de los procesos que transcurren espontáneamente, ya que el sistema evoluciona hacia un mayor desorden, lo que supone un aumento de entropía, ΔS>0.
Ambos casos los podemos resumir con la expresión:
Cuantitativamente, la variación de entropía de un sistema que pasa de un estado inicial a un estado final mediante un proceso reversible se define por:
Y si el proceso es irreversible:
donde en ambos casos ΔQ es cantidad de calor intercambiado por el sistema,
ΔS es la variación de entropía entre los estados inicial y final, y T es la temperatura absoluta a la que se produce la transformación.
Una mejor comprensión del concepto de entropía se la
debemos al físico austríaco Boltzmann.
Boltzmann acertó de pleno al proponer una interpretación
estadística de la entropía. En un momento en el que no estaba asentada la
teoría atomista de la materia sus ideas no fueron bien aceptadas cuando no
rechazadas. Quien sabe si fue este rechazo a sus ideas lo que lo llevó al
suicidio en 1906. Sus aportaciones fueron fundamentales para la teoría
cinética de los gases y del calor.
Aunque la termodinámica no hace suposiciones acerca de la estructura de la materia, puede mejorarse nuestra comprensión de las funciones termodinámicas si tratamos de interpretarlas en términos de las propiedades moleculares. Sabemos que la presión de un gas resulta de las colisiones moleculares con las paredes del recipiente, que la temperatura es un parámetro que expresa la energía cinética promedio de las moléculas, y que la energía interna consiste en las energías cinética y potencial de todos los átomos, moléculas, electrones y núcleos de un sistema.
¿Qué propiedad molecular refleja la entropía?
Existen dos maneras de describir el estado de un sistema termodinámico: la descripción macroscópica dada por los valores de las funciones de estado, tales como P, V y T, y la descripción microscópica que implicaría dar la posición y velocidad de cada átomo del sistema. Pensemos para un mol la cantidad tan inmensa de información que necesitaríamos, y esto sólo para un instante.
Cuando observamos cualquier sistema termodinámico en un estado de equilibrio macroscópico, su estado microscópico está cambiando a una velocidad enorme. A pesar de esta actividad molecular, las propiedades de un estado macroscópico permanecen constantes. Esto debe significar que existen muchos estados microscópicos compatibles con cualquier estado macroscópico.
La entropía es una medida del número de estados microscópicos asociados con un estado macroscópico particular.
Para explorar este punto utilicemos una baraja de cartas como un símil de un sistema termodinámico. Existen dos estados macroscópicos distintos de la baraja: o está "ordenada", con las cartas en alguna secuencia normal; o está "desordenada", con las cartas en una secuencia al azar. Podemos ver que sólo hay un estado microscópico "ordenado". Pero existen muchos estados microscópicos asociados con el estado macroscópico "desordenado", porque hay muchas secuencias al azar para las cartas. Puesto que la entropía mide el número de estados microscópicos del sistema, y aumenta con dicho número, podemos decir que el estado
"desordenado" tiene una entropía más alta que el estado "ordenado". Si
se nos caen las cartas al suelo y las recogemos rapidamente, lo más
probable es que las cartas estén desordenadas, pues hay más
combinaciones en las que las cartas estén desordenadas que combinaciones
en que las podamos considerar ordenadas. Para las cartas un estado
ordenado es menos probable que un estado desordenado.
Empleando este análisis podemos ver por qué una baraja de cartas cambia de un estado macroscópico
"ordenado" a un estado "desordenado" cuando las cartas son barajadas. Puesto que hay más estados microscópicos asociados con el estado macroscópico
"desordenado", para la baraja es simplemente más probable ir a parar a una situación de mayor
"desorden". Si aplicamos este razonamiento a la conducta de los sistemas termodinámicos, podemos ver que la entropía tiene una tendencia natural hacia el aumento, dado que esto corresponde al cambio de los sistemas desde las condiciones de baja probabilidad a los estados de mayor probabilidad.
Es cuestión de estadística.
¿Por qué estos estados "desordenados" son más probables que los estados
"ordenados"? La respuesta está en lo que realmente entendemos por "desorden". Un sistema desordenado es aquel del cual tenemos una cantidad relativamente pequeña de información referente a su estado microscópico exacto. La razón por la cual carecemos de este conocimiento detallado es que el sistema tiene muchos estados microscópicos asociados a él, y lo mejor que podemos hacer es suponer que en cualquier instante él está en alguno de los mismos. Si solamente fueran posibles unos cuantos estados microscópicos, podríamos ser capaces de hacer una conjetura exacta de aquel en el que estaba el sistema y, siendo así, hacer una descripción detallada de las posiciones y velocidades de las moléculas. De este modo, un sistema "desordenado" es aquel que tiene un número relativamente grande de estados microscópicos asociables a él, y esto es el porqué de que un estado desordenado sea más probable que un estado ordenado. Este
vídeo te puede ayudar a entenderlo.
Cuando hablamos de la entalpía encontramos útil seleccionar a un cierto estado de la materia y asignarle una entalpía de formación definida. Nuestra decisión de que la entalpía de formación de todos los elementos en sus estados estándar sea cero se basa solamente en la conveniencia. A cualquiera otro estado de los elementos se le podría asignar la entalpía cero. En el caso de la entropía el caso es diferente, porque la asociación de la entropía con el número de estados microscópicos disponibles para un sistema sugiere una selección natural de la entropía cero. En un cristal perfecto, en el cero absoluto, hay un solo estado microscópico posible. Cada átomo debe estar en un punto de la red cristalina y debe tener una energía mínima. Es así como podemos decir que éste es un estado de orden perfecto, o de entropía cero. Esta importante decisión es expresada por el
|
|
Tercer principio de la Termodinámica:
"La entropía de los cristales perfectos de todos los elementos y compuestos puros es cero a la temperatura del cero absoluto". |
|
La tercera ley nos permite asignar una entropía absoluta a cada elemento y a cada compuesto, a cualquier temperatura. Para un mol de
sustancia:
Si evaluamos esta integral desde 0K hasta 298K , para un mol de sustancia a una atmósfera de presión obtenemos la entropía estándar absoluta, Sº, que definimos como el valor de la entropía molar de una sustancia a la temperatura de 25ºC y presión de una atmósfera. Sus valores los encontramos tabulados, para las diferentes substancias, en J/molK.
Tabla
de datos químicos
|
|
ENTROPÍA DE REACCIÓN A
PARTIR DE ENTROPÍAS ABSOLUTAS
|
| La entropía es una función de estado, por lo tanto, la variación de entropía en una reacción química a temperatura constante se puede definir cómo:
Si expresamos esta ecuación en función de las condiciones estándar, 25ºC y 1 atm de presión:
La entropía estándar de reacción, de manera semejante a como hicimos con las entalpías, la podemos calcular a partir de los valores tabulados de las entropías absolutas.
EJEMPLO 3: Calcula la variación de entropía en la reacción:
CaCO3(s) → CaO(s) + CO2(g)
Dado que: ΔSºR = Σ np·Sºp − Σ
nr·Sºr
Utilizando la
tabla
de datos químicos.
ΔSºR = 1mol·Sº [CaO(s)] + 1mol·Sº [CO2(g)]
–
1mol·Sº [CaCO3] =
= 1mol (39,7JK–1mol–1) + 1mol (213,8JK–1mol–1)
– 1mol(92,9JK–1mol–1) =
160,6 JK–1
El valor positivo que se obtiene para la entropía en este ejercicio se puede predecir analizando el concepto de entropía. Para evaluar el signo de la entropía de un proceso se debe tener en cuenta que:
- Una reacción que origina un aumento en el número de moles de gas va siempre acompañada de un aumento de entropía. Si el número de moles de gas disminuye, ΔS será negativa.
- Si en una reacción no intervienen especies gaseosas, pero existe un aumento considerable en el número de moles de los productos respeto a los reactivos, también cabe esperar una ΔS positiva, tal es el caso de las sustancias hidratadas:
CuSO4·5H2O(s) →
CuSO4(s) + 5H2O(l) ΔS>0
pues se pasa de un mol en los reactivos a seis moles en los productos.
- Si el cambio en el número de moles no es muy significativo, podemos encontrar ΔS positivas y ΔS negativas, como por ejemplo:
2AgI(s) → 2Ag(s) + I2(s) ΔS = – 6,2
PbI2(s) → Pb(s) + I2(s) ΔS = 1,1
|
|
CRITERIO
DE ESPONTANEIDAD. ENERGÍA LIBRE DE GIBBS
|
| Una vez introducido el concepto de entropía estamos en condiciones de no confundir la tendencia a la realización de un proceso químico, con el calor intercambiado en el mismo, ya que además de este parámetro existe otro tan importante como el grado de desorden del sistema o entropía.
Existe una magnitud termodinámica que engloba y relaciona ambos parámetros, de una parte el calor intercambiado en el proceso,
ΔH, y de otra el desorden alcanzado en el mismo, ΔS.
Esta magnitud, ΔG, se llama energía libre de Gibbs o
entalpía libre y es, como ΔH y ΔS, una magnitud termodinámica extensiva y función de estado:
Empleando la energía libre de Gibbs es muy sencillo predecir la espontaneidad de los procesos.
Un proceso será espontáneo cuando ΔG sea negativo, es decir:
EJEMPLO 4: Calcula la variación de energía libre en la reacción siguiente en condiciones estándar:
CaCO3(s) → CaO(s) + CO2(g)
Calculando la ΔHºR y la ΔSºR a partir de los datos tabulados obtenemos:
ΔHºR = +177,7kJ/mol
ΔSºR = +160,6J/mol K
Utilizando la ecuación de Gibbs:
ΔGºR = ΔHºR –T·ΔSºR = 177,7kJ – 298K · 0,1606kJ/K = +129,8 kJ
Deducimos de este valor de la energía libre, ΔG>0, que la reacción no es espontánea a la temperatura de 25ºC.
Toda reacción que transcurra con merma de entalpía (ΔH<0) y aumento de entropía
(ΔS>0) será espontánea y la variación de energía libre, ΔG, siempre será negativa. De igual manera, aquellas reacciones en las que
ΔH sea positivo (endotérmicas) y ΔS sea negativo nunca serán espontáneas, pues
ΔG será siempre positivo.
Ahora bien, existen reacciones donde los términos entálpico y entrópico están enfrentados, y será la temperatura la magnitud que determine la espontaneidad o no del proceso. Observemos esto en un caso concreto, la descomposición del carbonato de calcio:
CaCO3(s) → CaO(s) + CO2(g) ;
ΔH >0
1. ΔH es positivo, lo que indica que la reacción es endotérmica.
2. Se pasa de una molécula en estado sólido a una molécula de sólido y otra de gas, con lo que el desorden aumentará,
ΔS positivo.
Como tanto ΔH como TΔS son positivos, el signo de la energía libre
ΔG = ΔH - TΔS dependerá del valor de T:
a) Si T es pequeño, ΔH será >TΔS, con lo que ΔG será positivo y el proceso nunca será espontáneo.
b) Si T es grande, ΔH será <TΔS, con lo que ΔG será negativo y el proceso será espontáneo.
c) Existirá un valor de T en el que se cumpla que ΔH = TΔS, con lo que
ΔG = 0 y el proceso se encontrará en equilibrio.
En el siguiente cuadro se presentan las diferentes alternativas:
| ΔH |
ΔS |
ΔG |
Observaciones |
| Negativa |
Positiva |
Negativa |
Espontánea a cualquier T, T no influye. |
| Positiva |
Negativa |
Positiva |
No espontánea a cualquier T. Ocurre el proceso inverso. |
| Positiva |
Positiva |
a T baja
Positiva |
A T baja no es espontánea, si ΔH>TΔS |
a T alta
Negativa |
A T alta la reacción es espontánea, si TΔS>ΔH. |
| Negativa |
Negativa |
a T baja
Negativa |
A T baja es espontánea, si |ΔH|>|TΔS| |
a T alta
Positiva |
A T alta no es espontánea, si |TΔS|>|ΔH| |
Recordemos por lo tanto que:
Si ΔG < 0 el proceso tiene lugar espontáneamente, evoluciona pasando de reactivos a productos.
Si ΔG > 0 el proceso no es espontáneo, y tiene lugar en sentido contrario pasando de productos a reactivos.
Si ΔG = 0 el proceso permanece en equilibrio coexistiendo reactivos y productos.
EJEMPLO 5: Calcula la temperatura a la cual esta reacción será espontánea:
CaCO3(s) → CaO(s) + CO2(g)
Sabiendo que ΔHºR = +177,7kJ/mol y ΔSºR = +160,6J/mol K en condiciones estándar, y suponiendo que estos valores no varían apreciablemente con la temperatura.
Si estos valores no varían apreciablemente con la temperatura podemos calcular a qué temperatura la reacción estará en equilibrio,
ΔG = 0,
Como el término de la entalpía es positivo y el de la entropía negativo, a temperaturas más altas de 833,5ºC el término de la entropía será mayor que el término de la entalpía y así la variación de energía libre será negativa, y por tanto la reacción será espontánea.
|
|
ENERGÍA
LIBRE NORMAL DE
FORMACIÓN
|
| Lo mismo que pasaba con las entalpías pasa con las energías libres, no podemos conocer sus valores absolutos. Pero podemos tabular los valores de las energías libres para las reacciones de formación de cada una de las sustancias que participan en una reacción química. Como la energía libre es una función de estado podremos calcular la energía libre de una reacción a partir de las energías libres de formación.
Definiremos la energía libre normal de formación de un compuesto en las condiciones estándar (T = 298K y P = 1atm) como el cambio de energía libre necesario para formar un mol de compuesto a partir de los elementos en estado fundamental en esas condiciones de presión y temperatura. Estos valores de
ΔGºf los tenemos tabulados para las principales sustancias. Al igual que pasaba con las entalpías las
ΔGºf de los elementos será cero por definición de energía libre de formación.
|
|
ENERGÍA
LIBRE DE REACCIÓN A
PARTIR DE ENERGÍAS LIBRES DE
FORMACIÓN
|
| Teniendo en cuenta que ΔG es una función de estado, que solo depende de los estados inicial y final, y que tenemos tabulados los valores de la energía libre normal de formación,
ΔGºf ,para diferentes compuestos, podemos calcular su variación en una reacción sin más que sumar las energías libres de los reactivos y productos que intervienen en la misma cuando ambos se encuentran en el estado normal, es decir, a la presión de 1atm si se trata de gases o a la concentración de 1mol/l para substancias en disolución líquida.
La variación de energía libre de una reacción química se define cómo:
Podemos concluir que ya estamos en condiciones de determinar si una reacción es espontánea o no, pero es conveniente distinguir entre espontaneidad y rapidez de una reacción química. Que una reacción sea espontánea significa que existe una tendencia natural para que la reacción se realice, pero puede que sea tan lenta, que en la práctica no se aprecie ningún cambio. Así, la combustión de la gasolina es espontánea, sin embargo, a menos que se aplique una chispa, la gasolina puede mantenerse en contacto con el aire durante largos periodos de tiempo sin que reaccione. Si una reacción es espontánea pero lenta, siempre es posible encontrar medios para acelerar el proceso, tales cómo elevar la temperatura o emplear un catalizador. Una reacción espontánea es una reacción
termodinámicamente posible. Si una reacción no es espontánea, como la inversa de la anterior (conversión de
CO2 y H2O en gasolina) jamás existirá un catalizador que la haga posible. Por lo tanto, el concepto de reacción espontánea o no espontánea permite al químico ver los límites de lo posible.
EJEMPLO 6: Calcula la variación de energía libre en la reacción siguiente en condiciones estándar a partir de datos tabulados de energías libres de formación:
CaCO3(s) → CaO(s) + CO2(g)
Utilizando la
tabla
de datos químicos.
ΔGºR = Σ np·ΔGºfp - Σ nr·ΔGºfr =
= 1mol·ΔGºf [CaO(s)]+1mol·ΔGºf
[CO2(g)] – 1mol·ΔGºf [CaCO3(s)]=
= 1mol·(–604,2kJ/mol) +1mol·(–394,6kJ/mol) –1mol·(–1128,8kJ/mol) = 130kJ
EJERCICIOS
PARA PRACTICAR
|
|
|
|